The truth about Ehlers Danlos Syndrome (EDS) Explained: What You Should Know
May is Ehlers Danlos Syndrome Awareness Month
It’s a great time to talk about Ehlers Danlos Syndrome because it’s awareness month for Ehlers Danlos Syndrome! Do you remember that guy on “Ripley’s Believe It or Not” long ago who was known for his insanely stretchy skin? His name is Garry Turner and he holds the Guinness World Record for the stretchiest skin since 1999. You guessed it, he has Ehlers Danlos Syndrome.
I’m not sure how he is these days, but aside from his stretchy skin, I’ll bet that he deals with symptoms that aren’t mentioned because his skin is what stands out the most to people. Underneath that skin, I’m sure he suffers from a myriad of symptoms that cause joint pain, subluxations/dislocations, GI issues, and much more. Maybe he doesn’t, or he just hides it very well, but either way, I’m going to break down the poorly misdiagnosed and misunderstood condition that needs more support and awareness.

What is Ehlers Danlos Syndrome?
EDS is short for Ehlers Danlos Syndrome, a group of inherited disorders that affects anywhere there is connective tissue in the body including blood vessels, skin, and joints. It’s considered a rare condition, but it doesn’t seem so rare. To me, it actually seems poorly misunderstood and often misdiagnosed. There are 13 known types with a 14th type, I hear that’s recently been discovered but has not yet been officially classified and named. It’s gonna be a long one, but stick around and I’ll explain more.
What causes EDS?
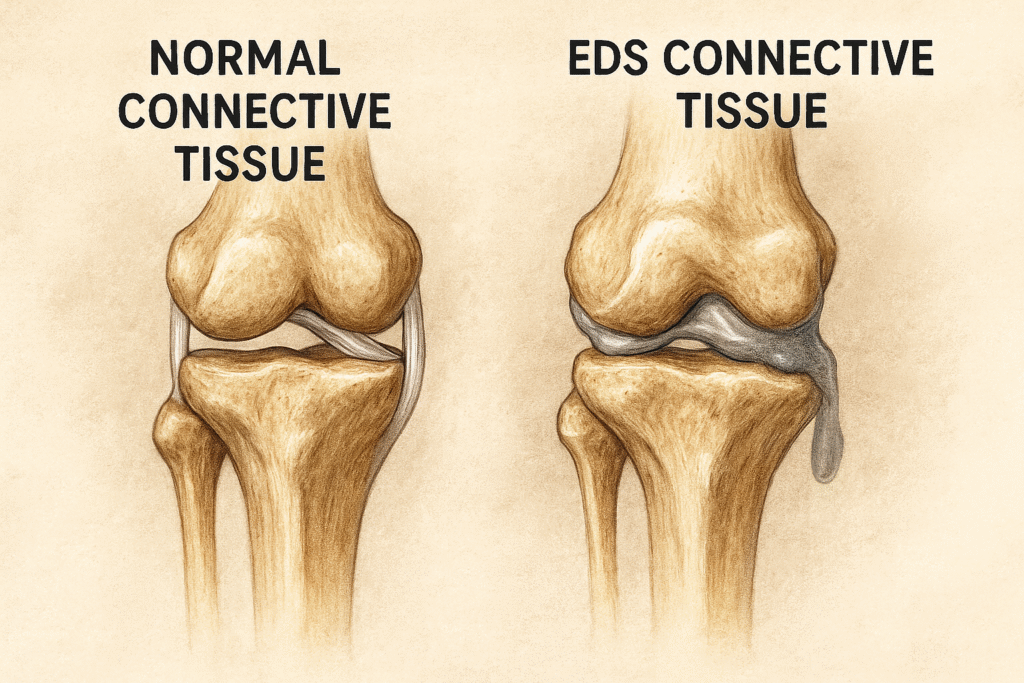
EDS is caused by genetic changes that affect the connective tissue. The Ehlers Danlos Society states, “The Ehlers Danlos syndromes are caused by changes in the genes that affect the structure and function of collagen and related connective tissue proteins.” Connective tissue is found all throughout the body, and its job is to protect, support, and provide structure to other parts of the body. You can think of it as the glue that holds the body together.
Living With EDS
Living with Ehlers Danlos Syndrome is life-changing and challenging on the daily. We are constantly labeled as hypochondriacs or judged, misdiagnosed, mocked, gaslit, and ignored. We are so much more than bendy joints and stretchy skin. Some of us are completely disabled from it, and we feel like we’ve lost a part of our lives. Many of us grieve our old lives before our symptoms became apparent and just fighting for answers is exhausting. A lot of us have families who rely on us to keep things running smooth at home, so most of the time we push our bodies beyond its limit and end up creating even more stress on the body.
Painful Movement
Our body’s range of motion is more than the average person’s due to the laxity of our joints caused by the weak connective tissue. We are met with a multitude of physical and emotional hurdles. Simple tasks like getting out of bed can be such a daunting task as our joints often feel stiff and painful, which makes moving painful and dreaded. Gripping a pen or pencil can be painful, and some of us rely heavily on ring splints to help stabilize our finger joints. We can even wake up with a hip or shoulder out of place just from sleeping. We truly never know what each day will bring us.
Heavy Fatigue
As the day goes on, dealing with chronic fatigue can really start to wear us down. It doesn’t take much—just making a meal, folding a basket of laundry, holding a cup of coffee can feel like a lot, or even a simple conversation can easily tire the voice and the body. Even taking a shower or doing basic self-care can be exhausting, and sometimes we need help to get through it.
Dysautonomia
Many of us with EDS also deal with some level of autonomic dysfunction (like POTS), which means our bodies don’t regulate things like heart rate, blood pressure, and temperature the way they should. Because of this, we often need to stay more hydrated than the average person just to feel somewhat “normal.” Electrolytes, extra fluids, and frequent water breaks become part of daily life and oftentimes intravenous fluids are needed. This is not because we’re being dramatic, but because our bodies genuinely need the extra support to function.
Muscle Weakness
Joint laxity and instability are common nuisances for us. Since the connective tissue that holds everything together is weakened, our necks struggle to keep our head upright, often leaving our heads feeling like a bowling ball that’s too heavy for our bodies, or most often described as a bobble head. This commonly leads to cranial cervical instability (CCI) and/or Atlanto-axial instability (AAI). Basically, that means there’s abnormal movement between the skull and the top part of the spine, which should be stable but isn’t.
Our body works overtime and our muscles overcompensate just to keep the head up, and because of that, the neck and shoulder muscles are constantly tight, sore, and worn out, leaving the head feeling unsteady. When you think about how important the spine is for everything, it makes sense that this kind of instability can cause a lot of neurological symptoms—nerve compression, dizziness, head pressure, nausea and so much more. Sadly, this is really common in people with EDS, but because of how subtle or complex it can be, it’s often missed or misdiagnosed without the right testing.

Why Dental Work Is So Challenging for People With EDS
Our jaws? Oh yeah… they love to pop out of place whenever they feel like it. So we have to be super careful with things like eating, yawning, and especially when having any dental work done.
And speaking of the dentist….whew, that’s a whole other discussion. Most of us with EDS don’t numb well with local anesthetics, due to our faulty collagen that’s loose, causing the numbing that’s injected to leak through the tissue instead of staying where it needs to numb the nerves. This can make dental visits a whole problem in itself, and I wish more dentists understood this.. I personally cannot handle Novocain as it makes my heart rate shoot through the roof, so I always have to ask for the version without epinephrine.
And because the numbing wears off fast, I usually need several injections just to get through one appointment… even then, sometimes I can still feel what they’re doing. This causes extreme anxiety for me, so my dentist will give me a mild sedative to help me relax before any work is done. I know I’m not the only one who experiences this.
We’re pretty much always aware of how we move… unless we forget for half a second. Because with EDS, you can literally partially dislocate (sublux) or even fully dislocate something just by picking something up or moving the wrong way.
What’s collagen got to do with it?
Let’s talk about our friend collagen…the stuff that’s basically responsible for keeping our skin looking full, smooth, and supported. It’s also the same thing that likes to disappear as we age… hello wrinkles.
There are actually 28 different types of collagen, and each one has its own job. Our bodies make collagen naturally, and it’s one of the main building blocks of connective tissue. So when collagen isn’t formed correctly (like with EDS), it affects way more than just our skin.
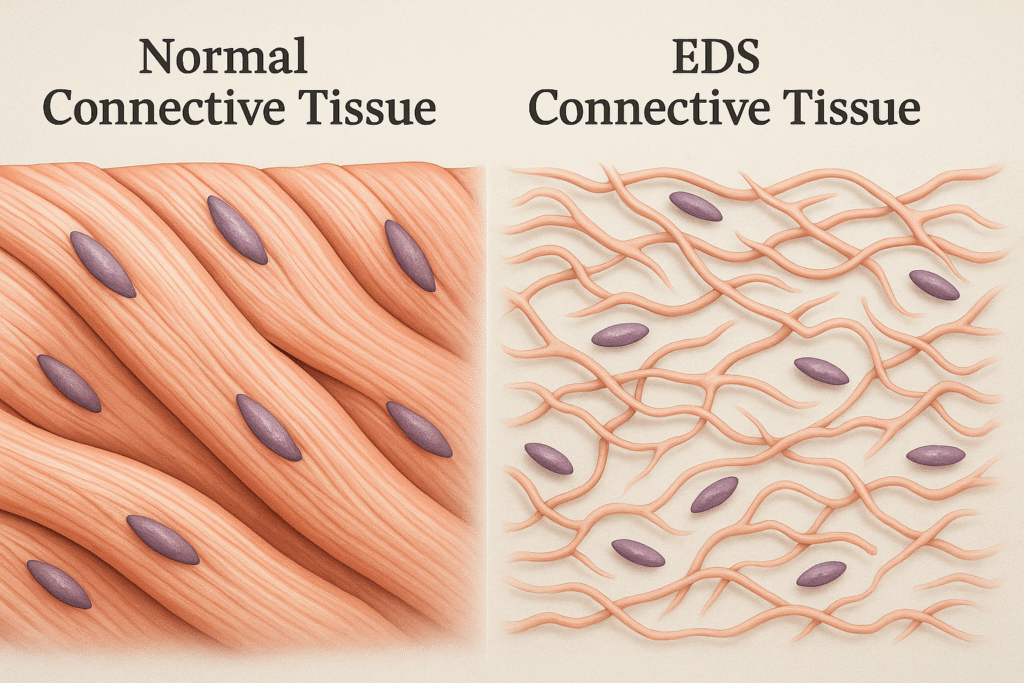
For those of us with Ehlers Danlos Syndrome, our collagen is basically faulty — which means it’s weaker than it should be. In a typical person, collagen is strong, supportive, and snaps back into place. I like to explain it as when you stretch a rubber band, and it immediately snaps back to its original shape.
But for someone with EDS? Our collagen behaves more like chewing gum or glue. It stretches… and stretches… and doesn’t snap back the way it’s supposed to. *I want to mention that not everyone with EDS will have skin as stretchy as mine.
Now imagine the inside of our bodies working like that. When collagen is made incorrectly, it affects everything — joints, skin, organs, and ligaments. That’s why we deal with hypermobility, unstable joints, soft or stretchy skin, easy bruising, chronic pain, and all kinds of internal fragility issues.
We’re definitely more than just ‘stretchy skin’ or ‘bendy joints’ — there’s a whole lot happening beneath the surface.


When Your Body Makes the Plans for You
Living with EDS means never fully knowing what our bodies are going to do from one minute to the next. And honestly, it’s not always the pain that makes going out or traveling difficult — it’s the GI issues, or the nauseating car rides, or the dizziness. When your stomach is unpredictable, you’re constantly worried about flare-ups, nausea, bloating, bathroom access, or suddenly feeling too sick to continue whatever you’re doing.
Aside from living with MALS (Median Arcuate Ligament Syndrome) and other commorbidities of EDS, my GI is so disrupted that I prefer to stay home comfortably close to my toilet. That alone creates so much anxiety, because even simple plans can feel risky. Add in the potential for sudden pain or joint instability, and it becomes even harder to feel confident saying yes.
Because of all this, many of us end up turning down social plans — not because we don’t want to go, but because we genuinely don’t know how our bodies will behave. Sometimes we push ourselves to go anyway, but we have to be prepared to leave early or step away if things suddenly take a turn. Unfortunately, our bodies just aren’t convenient for us.
So please keep inviting us. Please don’t take it personal if we decline. I’ll be the first to admit I’m not the most consistent friend — not because I don’t care, but because my body often decides for me. One thing this condition has taught me is to be prepared for anything… and to stop being so hard on myself when plans have to change. We really do try our best, but sometimes the most loving thing we can do — for ourselves and others — is to listen to our bodies.
What are the symptoms of EDS?
I previously talked about some of the symptoms experienced with the Hypermobile type of EDS but here are general symptoms for the many different types of EDS.
Each type of Ehlers Danlos Syndrome presents with a variety of symptoms and/or including:
- Joint hypermobility (our joints have a greater range of motion than usual)
- Joint/bone pain and fatigue due to increased mobility, joint instability, or dislocations and subluxations
- Stretchy skin that’s prone to tearing, muscle tearing, and easy bruising due to fragile capillaries that break easily under pressure
- Unusually soft velvety skin with cigarette paper-like skin
- Atrophic cutaneous scars
- Piezogenic papules on the heels of the feet
- Digestive issues such as bloating, nausea, constipation, slowed gut motility (gastroparesis), IBD (Inflammatory Bowel Disease), and Abdominal Vascular Compressions that affect digestion
- Heart problems, including arrhythmias or mitral valve prolapse
- Blue sclera of the eye
- Poor would healing
- Livedo Reticularis
- Muscle weakness
- Low bone density
- Organ rupture
- Abdominal vascular compressions such as MALS (Median Arcuate Ligament Syndrome), SMAS (Superior Messenteric Artery Syndrome, NCS (Nutcracker Syndrome), MTS (May Thurner Syndrome), PCS (Pelvic Congestion Syndrome)
- Poor dental health, despite proper care for teeth, such as gum disease, including gingivitis, periodontitis, is prone to cavities and more
- And other comorbidities such as Chiari Malformation, Cranial Cervical Instability (CCI) or Atlanto-Axial Instability (AAI), Tethered Cord Syndrome, Cerebrospinal Fluid (CSF) Leaks, Intracranial Hypertension (IIH), Dysautonomia such as Postural Orthostatic Tachycardia Syndrome (POTS), and more
This is not an all-inclusive list
13 types of Ehlers Danlos Syndrome
Ehlers Danlos Syndrome (EDS) is a family of genetic connective tissue disorders and there are currently 13 types of EDS, with a 14th being discovered. As mentioned before, each type has a lot in common but also displays very distinct signs and symptoms that vary in severity from person to person, including mild to life-threatening.
Some types are caused by gene mutations that affect the production of collagen, while other types may be caused by mutations in other genes. As each type may have varying levels of severity between individuals, managing care for someone with Ehlers Danlos Syndrome will depend on the individual symptoms they present with. I have to emphasize that no two cases will be the same, so just because someone presents with certain symptoms doesn’t necessarily mean the next person will present the same way.
It’s also important to rule out any other conditions that could mimic any of the types of EDS.
- Hypermobile EDS (hEDS)– This is the most common type of EDS and the most common features include generalized joint hypermobility, joint instability, smooth skin, chronic pain, and can have stretchy skin.
- Classical EDS (cEDS)– This type is much rarer than hEDS and common features include joint hypermobility, skin fragility with extensive atrophic scarring and very stretchy skin with velvety texture.
- Vascular EDS (vEDS)– This type is one of the most severe types and the symptoms and complications include arterial fragility with aneurysm, dissection, or rupture. It also involves organ fragility and rupture, extensive bruising, and pneumothorax.
- Periodontal EDS (pEDS)– This type is rare and its most distinguishing features include severe early onset of gum disease with tooth loss and pretibial plaques (discolored shins).
- Kyphoscoliotic EDS (kEDS)– This is also a rare type and involves congenital/early-onset congenital and congenital hypotonia.
- Spondylodysplastic EDS (spEDS)– Another rare type, and includes short stature, joint hypermobility, muscle weakness, limb bowing, and craniofacial features.
- Brittle Cornea Syndrome (BCS)– This type is rare and includes severe problems with the cornea of the eye that can lead to vision loss or blindness and severe hearing loss and more.
- Arthrochalasia EDS (aEDS)– This type is ultra-rare and the main features of aEDS include severe joint hypermobility, which can lead to frequent joint dislocations and sublaxations with chronic joint pain. Affected individuals may also have low muscle tone and scoliosis (a sideways curvature of the spine). Skin hyperextensibility and fragile skin are also common in aEDS, and individuals may be prone to easy bruising and slow wound healing.
- Musculocontractural EDS (mcEDS)– A rare type with main features of joint hypermobility, which can lead to joint dislocations and chronic pain, as well as characteristic facial features such as a small chin, a flat or broad nasal bridge, and large ears. Other symptoms may include developmental delay, vision problems, and hearing loss.
- Classical-like EDS (clEDS)– Another rare type with the main features of clEDS similar to those of classical EDS, but with some differences in the severity and frequency of symptoms. These may include joint hypermobility, skin hyperextensibility and fragility, easy bruising, and poor wound healing. Affected individuals may also have chronic musculoskeletal pain, scoliosis, and mitral valve prolapse, a condition where the valve between the heart’s left atrium and left ventricle doesn’t close properly.
- Dermatosparaxis EDS (dEDS)– This is a rare type with the main feature of being extremely fragile and loose skin, which may lead to easy bruising and poor wound healing. Affected individuals may also have joint hypermobility, hernias, and organ prolapse, as well as short stature, kyphoscoliosis, a curvature of the spine, and other skeletal abnormalities.
- Myopathic EDS (mEDS)– Another rare type that includes the main features of muscle weakness and decreased muscle tone, which can lead to delayed motor development and difficulty with motor skills. Affected individuals may also have joint hypermobility, scoliosis, and thin, fragile skin that bruises easily. In some cases, they may also have cardiomyopathy, a condition where the heart muscle becomes weakened and enlarged, and respiratory problems.
- Cardiac- valvular EDS (cvEDS)– Also a rare type of EDS with the main feature of cvEDS being heart valve problems, such as mitral valve prolapse, a condition where the valve between the heart’s left atrium and left ventricle doesn’t close properly and aortic root dilation, a condition where the aorta, the main artery that carries blood from the heart to the rest of the body, becomes enlarged. Affected individuals may also have joint hypermobility, thin, fragile skin that bruises easily, and hernias.
- ?- Currently being researched
You can learn more at EDS Types – The Ehlers Danlos Society.
When It’s Not EDS… It Might Be HSD
Hypermobility Spectrum Disorder (HSD) is a condition where a person has joint hypermobility along with symptoms like pain, instability, and frequent injuries — but they don’t meet all the criteria for Ehlers-Danlos Syndrome. People with HSD can also experience things like fatigue, headaches, digestive issues, and even symptoms of autonomic dysfunction (like dizziness or fast heart rate).
It’s still a real and valid connective tissue disorder. Whether someone has EDS or HSD, their experience is valid — and both conditions deserve awareness, support, and proper medical care.
How is EDS diagnosed?
Since Ehlers Danlos Syndrome is a genetic condition and being there are different types, they each have their own diagnostic criteria. Patients are usually referred to a geneticist or a rheumatologist who may be able to identify EDS. If a patient presents with symptoms during physical examination, a genetic test is usually done to determine if they have a certain gene of any of the EDS types except for the hypermobile type (hEDS)..
The genetic cause of Hypermobile EDS (hEDS) has not yet been identified but the Norris Lab is in collaboration with researchers from the Ehlers Danlos Society and others to find the gene associated with the hypermobile type of EDS. We are hopeful that a confirmed gene will be identified in order to diagnose hEDS through genetic testing in the future. This advancement could be life-changing for so many!
The average amount of time it can take to get a diagnosis is 10-12 years because the symptoms can mimic other conditions and not many doctors understand EDS. If you suspect you may have EDS, reach out to your doctor asap as it’s not always easy to get answers and you may be stuck waiting a while. I didn’t get answers until I was in my mid 20’s, eventhough I presented with symptoms as a child. And from my personal experience, if you don’t feel like you are getting any answers, it’s okay to look for another doctor. The Ehlers Danlos Society has an amazing page of resources that you can check out here Healthcare Professionals Directory – The Ehlers Danlos Society.
In the meantime, the diagnostic criteria for Hypermobile EDS include a full examination and must meet the diagnostic criteria which you can find here hEDS Diagnostic Checklist – The Ehlers Danlos Society . I personally don’t believe that the Beighton scoring criteria is not always suitable to diagnose hEDS as it doesn’t address all joints and can often miss a true diagnosis of hEDS. That has been a huge frustration for me. There is another alternative scoring criteria though that I was recently introduced to, which assesses other joints not mentioned on the Beighton score known as the Hospital Del Mar test. This could be brought up to your doctor if you suspect hEDS but fail the Beighton test.
Hopefully if and when the gene is discovered, it will help clear up a lot of the missed diagnoses of hEDS. It’s a struggle for us all trying to get answers. It would certainly make things easier for doctors and patients.
Treatment For EDS
There is no cure for EDS, but there are treatments that can help manage its symptoms, although not always. Each person’s care will be managed specifically for their symptoms. These may include physical therapy, medication, surgery, mobility aids, and/or equipment to help manage getting through the day, tube feeding, intravenous fluids, and more. Usually, a person’s care will consist of a team of experts who manage the different areas of care including cardiology, neurology, orthopedic, pain management, and more. What works for one person may not work for the next. Life with EDS can get pretty overwhelming and exhausting while keeping up with the care that comes with it, but we do our best!
Embracing Strength, Inspiring Resilience as a Zebra
With all that said, we just have some messed up genes. I’m kidding, well… we kind of do, but we’re pretty cool. I mean, if you haven’t met someone like us yet, hello! We’re called Zebras and here’s why.
Medical students are taught in Med school to think hoof beats like a horse and not to expect to see a zebra. In essence, this means that doctors should consider the most typical cause of a patient’s symptoms rather than the more unusual ones. We are the unusual ones, we are the unexpected cases! That’s why we’re called Zebras 🦓
Check out this episode from The Resident that highlights Ehlers-Danlos Syndrome:
👉 Watch The Resident Clip and Grey’s Anatomy — EDS Scene

“Sometimes when you hear hoofbeats, it really is a zebra.”
Living with EDS is far more than the stretchy skin and bendy joints people see online. It’s a daily balancing act, a constant learning curve, and honestly, a battle most people never realize we’re fighting.
Ehlers Danlos Syndrome is complex, misunderstood, and often invisible — but the more we talk about it, the more awareness and understanding we create. My hope is that this post helps you better understand what EDS really is, how it affects daily life, and why early recognition matters.
Whether you’re someone living with EDS, suspect you might have EDS, supporting a loved one, or simply wanting to learn — thank you for taking the time to read and educate yourself. Awareness is one of the most powerful tools we have, and your willingness to understand this condition truly makes a difference!
For my zebra friends, I hope this has helped you feel seen and a little more understood. You’re not alone in this. ❤️🦓
Let’s Keep In Touch
I created this space because I spent years feeling unseen, unheard, and misunderstood.
If this post made you feel even a little more validated, I’d love for you to stick around and be part of this little Spoonie corner of the internet.
💌 Email Community
Get gentle reminders, cozy encouragement, Spoonie-friendly tips, and updates from my own journey.
Sign up below — I’d love to send support right to your inbox.

🎥 TikTok: @spoonie_girl
▶️ YouTube: @OutofSpoonsWithAmanda
Join me for real-life Spoonie moments — the honest days, the hard days, and the hopeful ones.
🙏 Need prayer?
If you’re walking through something heavy, you’re welcome to reach out.
I would love to pray for you. My faith has got me through some of the hardest parts of my life and I will continue to rely on it!
Send me your prayer requests, unspoken or spoken ❤️
📨runningoutofspoons1@gmail.com
Gentle Hugs,
Amanda
References:
https://rarediseases.info.nih.gov/diseases/6322/ehlers-danlos-syndromes